Distinct Classes of Complex Structural Variation Uncovered across Thousands of Cancer Genome Graphs.
October 1, 2020
Abstract
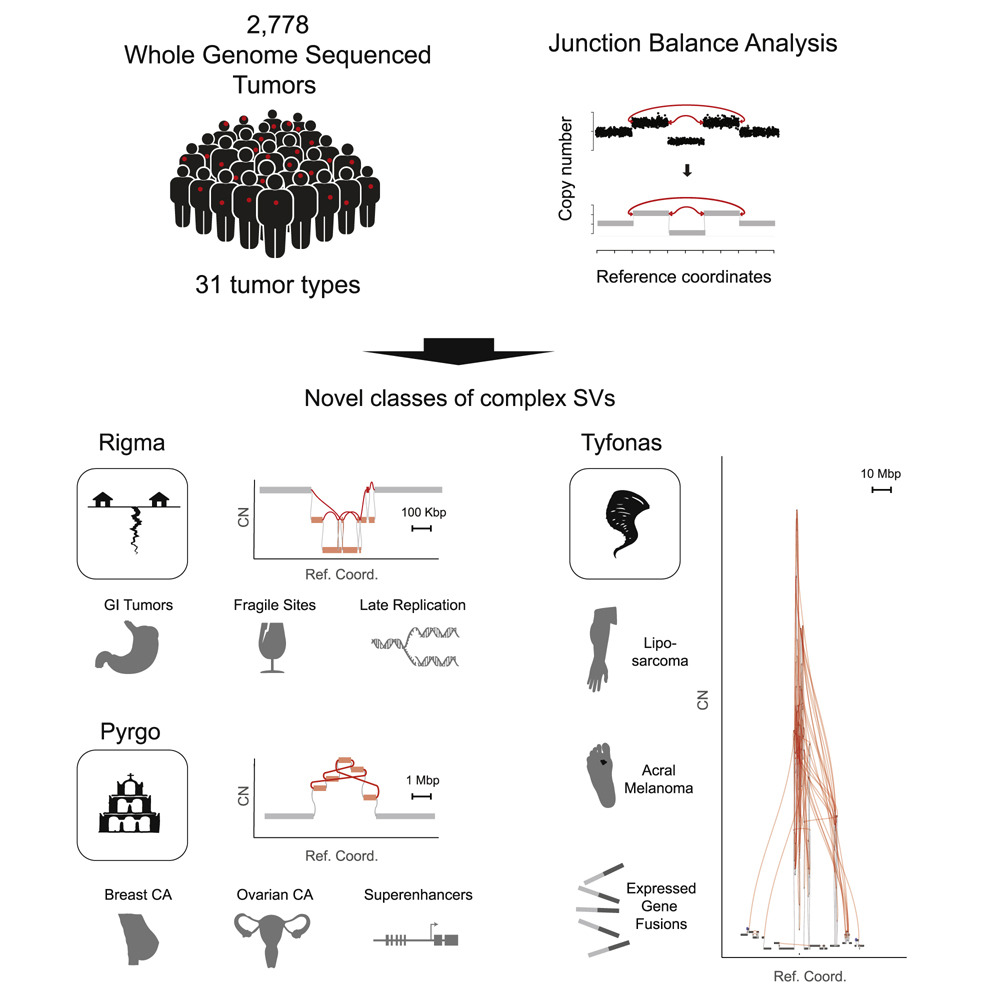
Cancer genomes often harbor hundreds of somatic DNA rearrangement junctions, many of which cannot be easily classified into simple (e.g., deletion) or complex (e.g., chromothripsis) structural variant classes. Applying a novel genome graph computational paradigm to analyze the topology of junction copy number (JCN) across 2,778 tumor whole-genome sequences, we uncovered three novel complex rearrangement phenomena: pyrgo, rigma, and tyfonas. Pyrgo are “towers” of low-JCN duplications associated with early-replicating regions, superenhancers, and breast or ovarian cancers. Rigma comprise “chasms” of low-JCN deletions enriched in late-replicating fragile sites and gastrointestinal carcinomas. Tyfonas are “typhoons” of high-JCN junctions and fold-back inversions associated with expressed protein-coding fusions, breakend hypermutation, and acral, but not cutaneous, melanomas. Clustering of tumors according to genome graph-derived features identified subgroups associated with DNA repair defects and poor prognosis.